fetching data ... 
Background: The treat-to-target approach and the availability of various targeted therapeutic agents have significantly advanced the treatment of rheumatoid arthritis (RA). However, therapeutic response to different medications still cannot be predicted, making it difficult to choose the right therapeutic agent in time. Therefore, understanding the pathophysiological processes of RA and the mechanisms of action of different therapeutic agents is crucial today. Interleukin-6 (IL-6) is a key inflammatory cytokine in the pathomechanism of RA, functioning through Janus kinase (JAK) 1, JAK2, and Tyrosine kinase (Tyk)2 kinases. It induces the phosphorylation of the signal transducer and activator of transcription (STAT) 3 protein, which initiates gene expression processes that mediate the biological effects of IL-6. Several targeted therapies act via the IL-6 and JAK-STAT pathway.
Objectives: The primary objective of our research was to develop an experimental model to evaluate the activation and inhibition potential of the JAK-STAT signaling pathway in RA patients and healthy controls (HC) in order to investigate the pathomechanism and treatment responses in RA more thoroughly.
Methods: In our research, we are examining the IL-6/JAK1-JAK2-Tyk2/STAT3 signaling pathway using flow cytometry by measuring the levels of phosphorylated STAT3 (pSTAT3). Peripheral blood samples were collected from RA patients and healthy controls (HC). Total blood samples were incubated with IL-6 and clinically available JAK inhibitors (tofacitinib, baricitinib, upadacitinib, filgotinib). Leukocytes (T cells, B cells, NK cells) were labeled with surface markers (anti-CD3, anti-CD19, anti-CD56) and anti-pSTAT3 antibodies to determine intracellular pSTAT3 levels, using the Beckman Coulter PerFix EXPOSE (Phospho-Epitopes Exposure) intracellular staining kit. The prepared samples were measured with a CytoFLEX (Beckmann Coulter) flow cytometer. Data was analyzed with CytExpert Software and GraphPad Prism10.
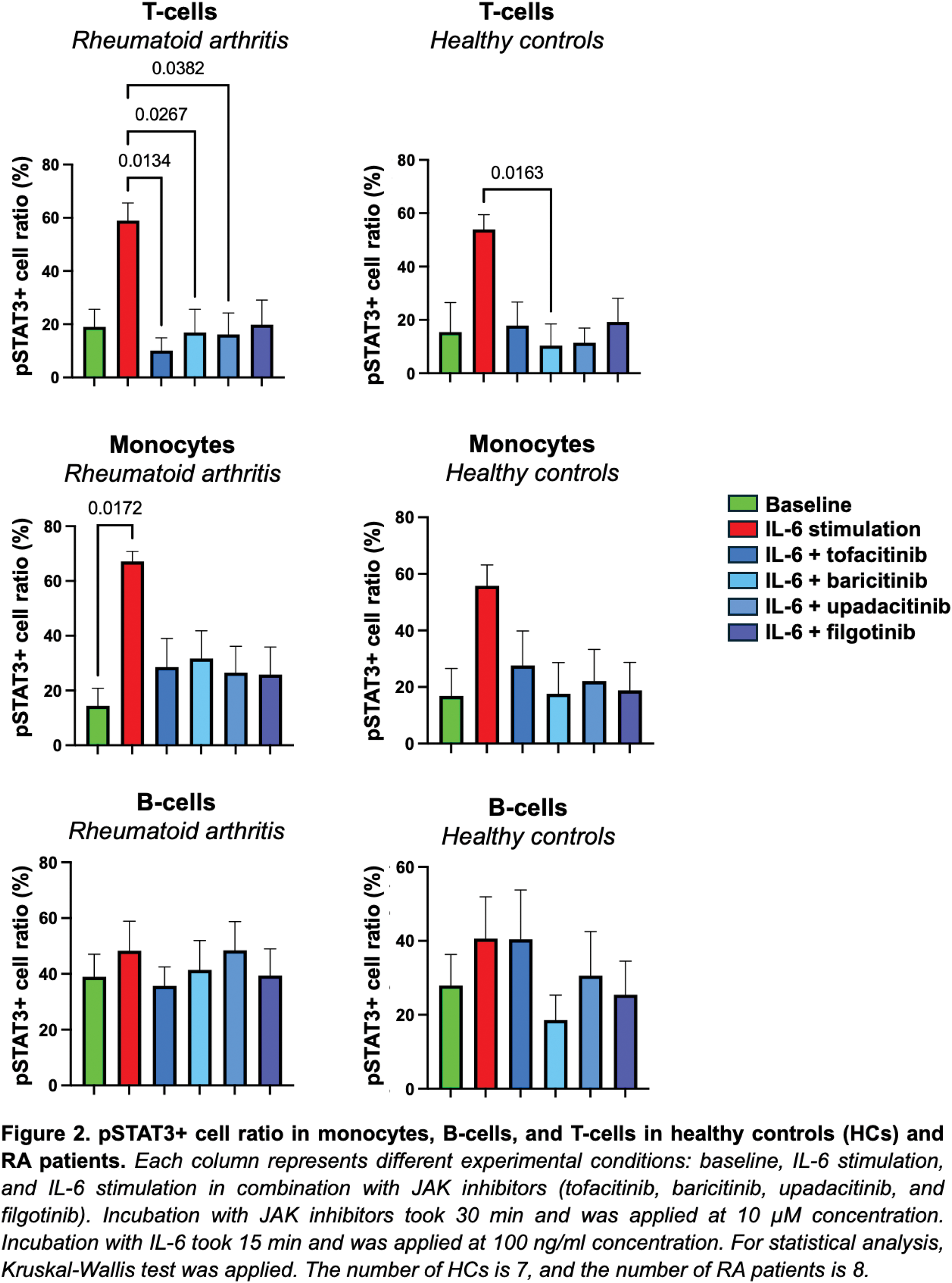
Results: Initially, we determined the optimal IL-6 stimulation concentration and incubation time for sample preparation. IL-6 concentrations of 1 ng/ml, 10 ng/ml, 100 ng/ml, and 1000 ng/ml were tested with incubation times of 15 minutes, 30 minutes, one hour, two hours, and twenty-four hours. Based on serial flow cytometry measurements, the highest pSTAT3 levels were observed with a 100 ng/ml IL-6 concentration. 15 minutes of incubation time was proved to be the most efficient. We further examined the optimal concentration (0.1 µM, 1 µM, 10 µM) and incubation time (30 minutes, 60 minutes) for JAK inhibitors (tofacitinib, baricitinib, upadacitinib, filgotinib) in the HC blood samples. The greatest reduction in pSTAT3 levels compared to control for all four JAK inhibitors was observed with a 10 µM concentration and a 30-minute incubation time (Figure 1). In this ongoing study, we are collecting samples from HCs and RA patients. To date, we have analyzed the data of 8 RA patients and 7 HCs. Figure 2 shows the percentage of pSTAT3+ cells in monocytes, B-cells and T-cells from both control and RA groups. Each column represents different experimental conditions: baseline, IL-6 stimulation, and IL-6 stimulation in combination with JAK inhibitors (tofacitinib, baricitinib, upadacitinib, and filgotinib). IL-6 stimulation significantly increased the pSTAT3+ monocyte ratio in RA patients (p = 0.0172), and the pSTAT3+ T-cell ratio was substantially reduced by baricitinib (p = 0.0163) in HCs and by tofacitinib (p = 0.0382), baricitinib (p = 0.0267) and upadacitinib (p = 0.0134) in RA patients.
Conclusion: In summary, we were able to set up an experimental model, which allows us to study the activatability and inhibiting potential of JAK-STAT signaling pathways. Based on data from measurements on total blood samples, the most significant pSTAT3 signal was observed with a 100 ng/ml IL-6 concentration, which could be inhibited by adding a 10 µM concentration of different JAK inhibitors. Further goals include increasing the number of healthy and RA control samples for testing.

REFERENCES: NIL.
Acknowledgements: This research was supported by the National Research, Development, and Innovation Office’s Thematic Excellence Program (TKP2021-EGA-29). LGT received the Richter Talentum Foundation PhD Scholarship.
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (