fetching data ... 
Background: Mitochondria are essential eucaryotic cell organelles with bacterial features, such as a double-stranded circular genome (mtDNA) with hypomethylated CpG areas. A fundamental role of mitochondria in autoimmunity was recently demonstrated. In brief, mitochondrial ROS participate in the formation of neutrophil extracellular traps (NETs), while extrusion of cell-free mitochondria and highly oxidised interferogenic mitochondrial DNA (ox-mtDNA) causes autoimmune disease in an animal model. In other connective tissue diseases, plasma mtDNA is a diagnostic biomarker, also reliable in the monitoring of disease activity, while platelets might also contribute as a major source of circulatory mtDNA.
Objectives: The present study aimed to explore the proinflammatory role of extracellular mtDNA in systemic sclerosis (SSc) and the cellular mechanisms controlling its release (SSc).
Methods: Total DNA was isolated from plasma samples of healthy controls (HC) and SSc patients. Mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) copy numbers in the plasma were measured by qPCR. mtDNA was isolated from HC and SSc patient platelets. Neutrophils and platelets were isolated from EDTA and citrate blood, respectively; cells were incubated with SSc patients’ plasma and mtDNA, and NET formation was assessed by SytoxGreen staining. Platelets were tested for mtDNA release propensity. DNA oxidation was evaluated by MitoSOX Red staining in vitro and 8-OHdG ELISA of patient plasma. Plasma IFN type 1 was measured by ELISA. Platelet activation was assessed by IF staining in vitro and ELISA for CXCL4.
Results: mtDNA levels were nearly 100-fold elevated in SSc plasma (33 consecutive unselected SSc patients) compared to age and sex-matched HC plasma (n=33) while nDNA plasma levels did not differ. SSc plasma enhanced NET formation and mtDNA release from HC neutrophils better than HC plasma, this feature of plasma was abolished by DNase1 preincubation. The NETs contained oxidized mtDNA. SSc platelets also contained and released oxidized mtDNA in conjunction with CXCL-4. mtDNA isolated from SSc platelets, promoted its own release by NET formation and by platelet activation more efficiently than HC-derived mtDNA. Moreover, mtDNA amounts correlated with CXCL4 and type I IFN levels in SSc plasma. SSc plasma-derived mtDNA was more potent than HC-derived mtDNA in triggering the IFN pathway in THP1 reporter monocytes, a process that was dependent on TLR9, JAK-STAT and cGAS-STING signaling. The type I IFN pathway, in turn, promoted NETosis, as confirmed by the use of JAK inhibitors on neutrophils.
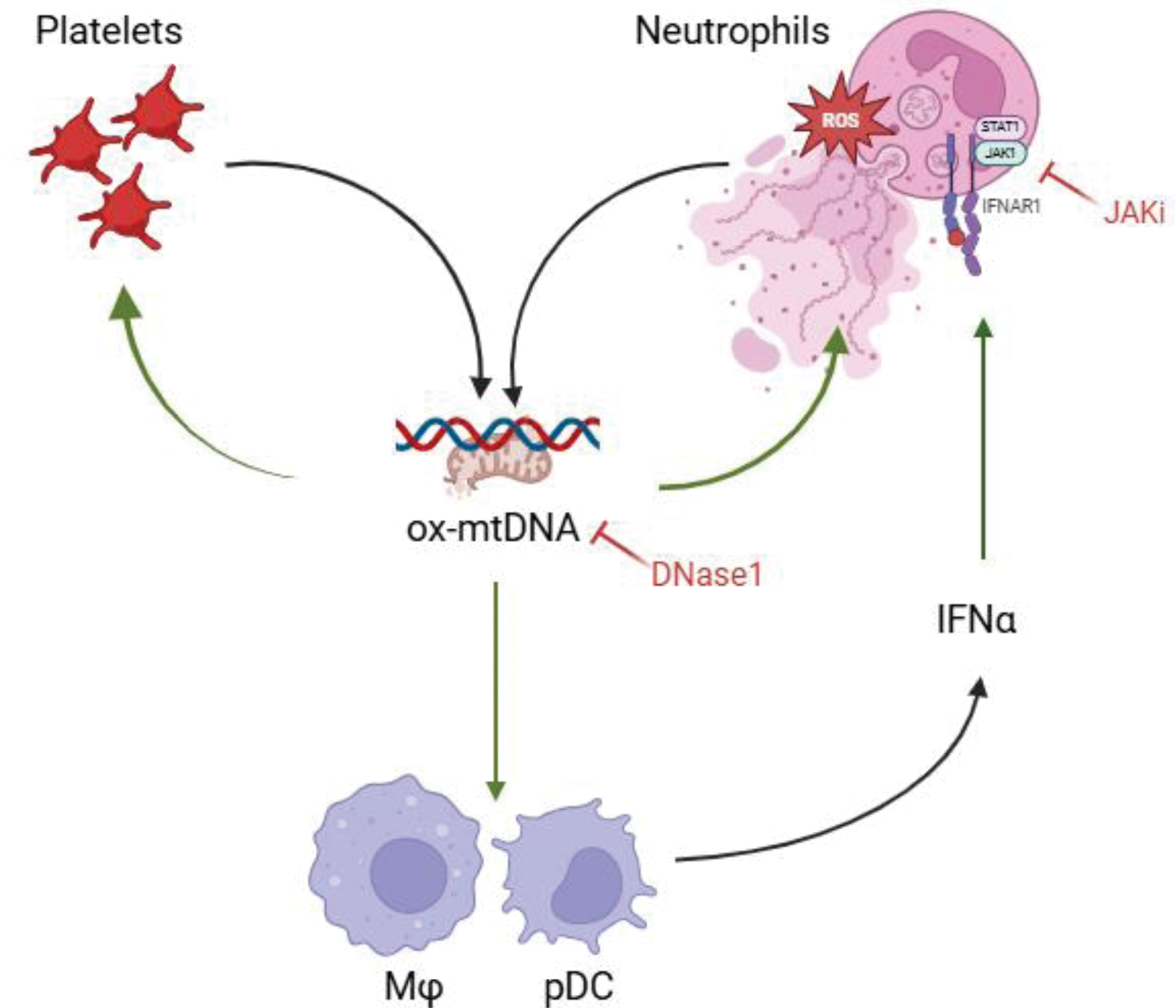
Conclusion: SSc plasma is characterized by an abundance of mtDNA that promotes positive feedback loops driving its own release by NETs and platelets. Moreover, mtDNA is particularly interferogenic and may contribute to tissue damage and fibrosis in SSc (Figure 1).
REFERENCES: NIL.
Schematic representation of the feedback loop involving oxidized mtDNA, released from activated platelets and NETotic neutrophils, which amplifies inflammation during SSc pathogenesis. SSc neutrophils and SSc platelets are poised to release ox-mtDNA. SSc mtDNA in turn promotes its own release by excessive NET formation from HC neutrophils and platelets. SSc platelets are in an activated state and release high levels of CXCL4, a confirmed biomarker of SSc. The oxidized nature of this mtDNA renders it prointerferogenic. SSc blood cells are characterized by a pronounced ISG, while IFNa1 levels are elevated in SSc patients’ plasma. mtDNA copy numbers correlate with IFNa1 levels. DNase1 pre-treatment abolishes mtDNA-release and mtDNA oxidation induced by SSc plasma. Finally, IFN pathway blockade significantly inhibits NET formation and mtDNA oxidation.
Acknowledgements: NIL.
Disclosure of Interests: Stavros Giaglis: None declared, Diego Kyburz Not relevant for this abstract, Not relevant for this abstract, Not relevant for this abstract, Ulrich A. Walker Not relevant for this abstract, Not relevant for this abstract, Not relevant for this abstract.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (