fetching data ... 
Background: Granulomatous diseases present notable diagnostic challenges. This case describes a rare mass-forming variant of ANCA-associated vasculitis (AAV), initially misdiagnosed as Erdheim-Chester Disease (ECD) due to foamy histiocytes. The mass-forming AAV variant, which can mimic malignancies or other conditions, is rarely documented in multi-organ involvement. This case highlights the need for advanced diagnostics and interdisciplinary collaboration to overcome delays and ensure proper treatment.
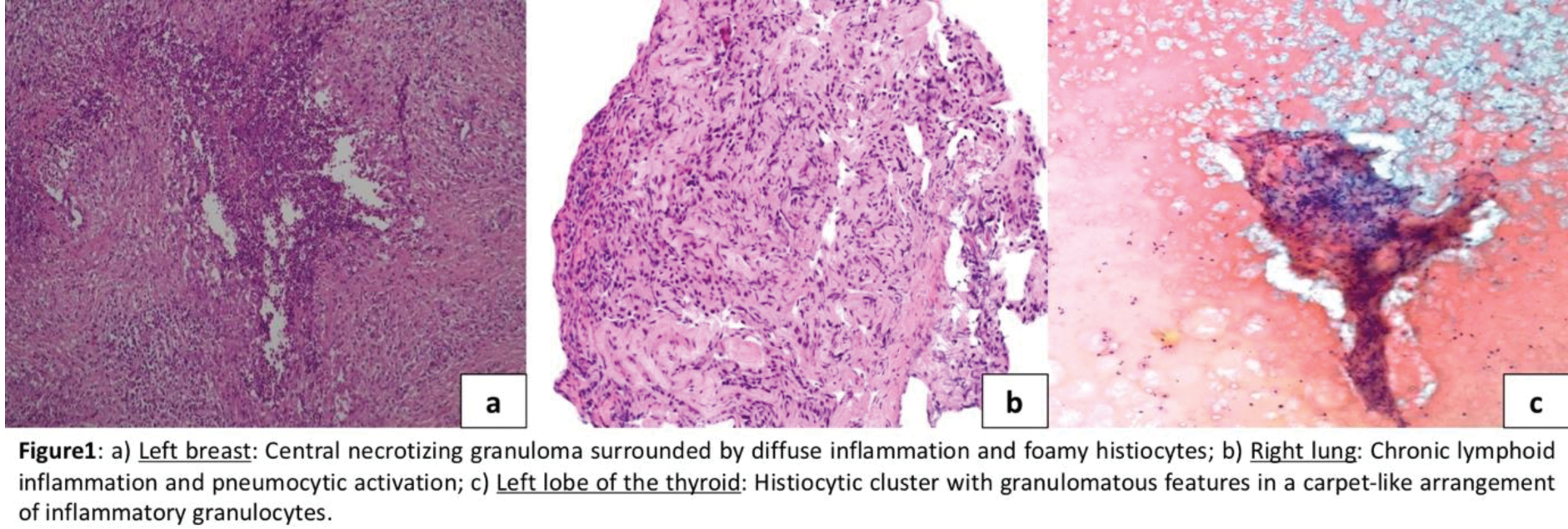
Case presentation: In 2018, a routine mammogram showed a breast lesion in a middle aged woman, which, upon histological analysis following a quadrantectomy, revealed chronic granulomatous inflammation characterized by the presence of foamy histiocytes [Figure 1a]. In 2021, the patient developed fever, elevated inflammatory markers, and pulmonary consolidations. Lung biopsies also revealed granulomatous inflammation with foamy histiocytes, prompting empirical steroid therapy [Figure 1b]. A subsequent thyroid ultrasound revealed a hypoechoic lesion, which was further investigated through fine-needle aspiration and found to exhibit histiocytic inflammation [Figure 1c]. Given the recurrence of granulomatous lesions in the lungs, breast, and thyroid, a histochemical re-evaluation for CD68 was requested, which returned positive. Consequently, a diagnosis of ECD was established, and interferon (IFN) therapy was initiated. Despite the ongoing therapy, systemic inflammation was persistent, and the patient experienced recurrent symptoms, such as fever, fatigue, and inflammatory arthralgia while ANCA and autoantibody profiles were negative. A re-evaluation of prior findings excluded ECD, leading to the interim diagnosis of “Granulomatous Disease Not Otherwise Specified” and the empirical initiation of cyclosporine and methotrexate. In 2023, the patient developed new pulmonary consolidations along with epistaxis and haemoptysis. Retesting revealed a high anti-PR3 ANCA titer, confirming a revised diagnosis of PR3-positive GPA involving the lungs, thyroid, and breast and ENT system. Induction and maintenance therapy with rituximab, and gradual steroid taper resulted in a quick and persistent remission.
Learning points for clinical practice: This case emphasizes the diagnostic difficulty of granulomatous diseases, especially when uncommon features like foamy histiocytes are present, leading to misdiagnoses and treatment delays. The rarity of mass-forming ANCA-associated vasculitis further complicates diagnosis, as it often mimics malignancies or other inflammatory conditions. Accurate diagnosis of rare diseases like mass-forming GPA requires a multidisciplinary approach, combining clinical insights, advanced diagnostics, and repeated histopathological assessments for effective management.
REFERENCES: NIL.
Acknowledgements: NIL.
Disclosure of Interests: None declared.
© The Authors 2025. This abstract is an open access article published in Annals of Rheumatic Diseases under the CC BY-NC-ND license (